
X射线光电子能谱(X-Ray Photoelectron Spectroscopy, XPS),在物理学上与俄歇电子能谱(AES)同根同源,两者常常能在同一台仪器上实现。XPS利用特征X射线激发固体,使其发射出光电子,这些电子的能量由高能量分辨率的能谱仪进行分析——这套能谱仪通常也用于AES分析。因此,许多XPS仪器会额外配置一个电子枪以兼容AES功能。XPS的X射线源通常是Mg Kα或Al Kα射线,而在更先进的仪器中,则普遍采用单色化的Al Kα X射线。
其核心物理过程是,能量为hν的X射线光子直接将固体中的内层电子(核电子)激发出来,这些出射的光电子具有的动能E可以由下式给出:
E = hν - E₁
这个公式是XPS技术的基石。我们真正关心的,正是这个E₁值,即内层电子的结合能。它像一个指纹,携带着所分析元素的化学状态信息。在XPS中,E₁通常取正值,并且能谱图直接使用结合能作为标尺,而非像AES那样参考费米能级的动能。关于不同元素的结合能数据,可以在各类手册、教科书和专业网站上查阅。一个需要注意的细节是,XPS和AES在能级命名上存在差异。AES常沿用X射线命名法,而XPS则很少如此。在XPS领域,我们采用能级数、亚层字母和自旋轨道耦合数来精确描述,例如,AES中的Mv或M₅在XPS中对应的是3d₅/₂。
在初始的光电离过程之后,原子内部会留下一个内层空穴。这个空穴可以通过俄歇过程被填补,并伴随一个俄歇电子的发射。这个俄歇电子同样会出现在最终的能谱图上,成为一个需要识别的特征峰。
XPS分析中产生的光电子,其动能大多落在500至1500 eV的区间内,这决定了XPS具有与AES相似的表面灵敏度。对于一些关键元素,其特征峰的动能甚至可以低至200 eV。
XPS的样品处理流程大体上与AES相似,但其分析对象的类型却常常大相径庭。XPS经常用于分析环境污染物,如聚二甲基硅氧烷(PDMS)及其类似物,因此样品通常很少进行清洁处理。
一个更常见的挑战是,XPS分析的样品更多是绝缘体,这就对荷电控制与荷电校正提出了极高的要求。ISO 19318标准中给出了大多数重要的处理方法,但具体能达到何种效果,很大程度上取决于仪器的具体配置。XPS能够分析聚合物薄膜或粉末样品,而这些通常不是AES的研究对象。
在某些仪器上,将样品置于金属网格或小孔下,是一种有效的荷电控制方法;但在另一些仪器上,这样做反而会导致谱峰展宽。对于配备了单色化X射线源的系统,可能需要电子中和枪或极低能量的离子流来进行荷电控制。无论采用何种方法,分析人员必须确保样品不会受到不必要的辐射损伤,因为这类样品极易因热、电子或离子的轰击而发生降解。
与AES类似,XPS的能量标尺校准通常使用溅射清洁后的铜(Cu)和金(Au)标准样品,并设定其发射角≤ 56°。测量得到的特征峰能量会与表1中的参考值进行比对。校准时,峰值能量取自原始谱图的峰顶,不进行任何背景扣除。
表1. 结合能标尺上特征峰位置的参考值 Erefn
| 峰编号 n | 归属 | Erefn (eV) |
|---|---|---|
| Al Kα | ||
| 1 | Au 4f7/2 | 83.95 |
| 2 | Ag 3d5/2 | |
| 3 | Cu L3VV | 567.93 |
| 4 | Cu 2p3/2 | 932.63 |
在表1中,3号峰是一个俄歇电子峰。对于非单色化的X射线源,这个峰的校准效果很好。但对于单色化系统,情况就变得复杂了。非单色化X射线源的Kα₁和Kα₂射线的线型与能量在所有仪器上基本一致,然而,经过单色器处理后的Kα射线,其线型和能量会因单色器的设置及其热稳定性而产生显著变化。通过调整单色器设置,甚至可以在不太损失强度的情况下,使测得的峰位在0.4 eV的动能范围内移动。因此,对于单色化系统,表1中的Cu俄歇峰被替换为银(Ag)的3d₅/₂光电子峰,这意味着校准时需要额外制备一个洁净的Ag样品。
ISO 15472标准将表1的校准方法整合进一个完整的协议中,涵盖了校准流程、不确定度评估、公差限设定以及校准周期的制定。对于在质量体系下运行的实验室,以及力求数据有效性的分析人员来说,遵循这些规程至关重要。使用该标准对一台维护良好的现代能谱仪进行校准,通常可以达到±0.2 eV的公差(四个月内)或±0.1 eV的公差(一个月内),之后才需要重新校准。对于大多数应用场景,±0.2 eV的精度已经足够。
XPS强度标尺重复性的评估与AES相似。其中,探测器的性能,特别是ISO 21270中关于强度标尺线性度的规定,具有决定性意义。在XPS中,强度比率是通过测量洁净铜样品的Cu 3p和Cu 2p₃/₂峰面积来确定的,测量前需要扣除一个Shirley背景。对用于确定Shirley背景的端点进行平滑处理,可以提高精度,使一系列测量中的重复性达到0.2%的优异水平。ISO 24237则详细描述了如何从铜样品中获得高质量数据所需的信号水平和操作流程,以及如何将其构建到监控协议中,并为控制图设置公差限,以确保强度测量始终满足应用要求。
一个令人遗憾的事实是,不同实验室间的光谱比对研究表明,所获得的谱图形状存在显著差异。如果所有实验室都采用相同的相对灵敏度因子(RSF),这种差异会导致定量结果出现高达两倍的偏差。
这里的症结、基本原理以及评估和使用仪器响应函数(IERF)的方案,与前面讨论的AES强度标尺校准完全一致。唯一的区别在于,我们不再使用5 keV的电子束来测量Cu、Ag和Au参考箔,而是使用Al或Mg的X射线,其入射角相对于表面法线在0°到50°的范围内。
虽然可以从各种手册、教科书和出版物中找到相对灵敏度因子,但早期的灵敏度因子数据差异巨大。即便是后来的数据,是否与其目标仪器兼容,或者是否仍然存在显著的不确定性,都还是未知数。自1986年的一次评估显示灵敏度因子数据集非常不稳定以来,一直没有进行过新的系统性分析。造成这种混乱的部分原因是,这些手册在制定数据时并未测量所用仪器的IERF。
沿用AES的分析流程,对于纯元素A的亚层Xᵢ,单位能量为hν的光子在立体角dΩ内产生的光电子强度I_AXi^∞由下式给出:
I_AXi^∞ = n_AXi σ_AXi secα N_A Q_A(E_AXi) λ_A(E_AXi) × [1 + 1/2 β_effAX (3/2 sin²γ - 1)] × cosθ (dΩ/4π)
其中,n_AXi是A元素X壳层i亚层中的电子布居数,σ_AXi是该内层电子对能量为hν的光子的电离截面,α是X射线束相对于表面法线的入射角,γ是入射X射线束与光电子出射方向之间的夹角,其余项与AES中的定义相同。n_AXi和σ_AXi的乘积值通常取自Scofield的数据,其他截面数据已被证明准确性较低。
当γ = 54.7°时,这个角度被称为“魔角”,此时上式方括号中的项等于1。但在其他角度,当γ > 54.7°时,该项通常大于1。参数β在气相分析中有效,但在固体中,Jablonski的研究表明,由于弹性散射的影响,β会衰减为有效值β_eff。Seah和Gilmore将Jablonski的蒙特卡洛数据简化为如下一组方程:
β_eff(θ) = β_eff(0) (1.121 - 0.208 cosθ + 0.0868 cos²θ)
以及
β_eff(θ) / β_eff = 0.876 [1 - ω (0.955 - 0.0777 lnZ)]
这里的ω值可以从相关图表(如图1)或数据库中查得。
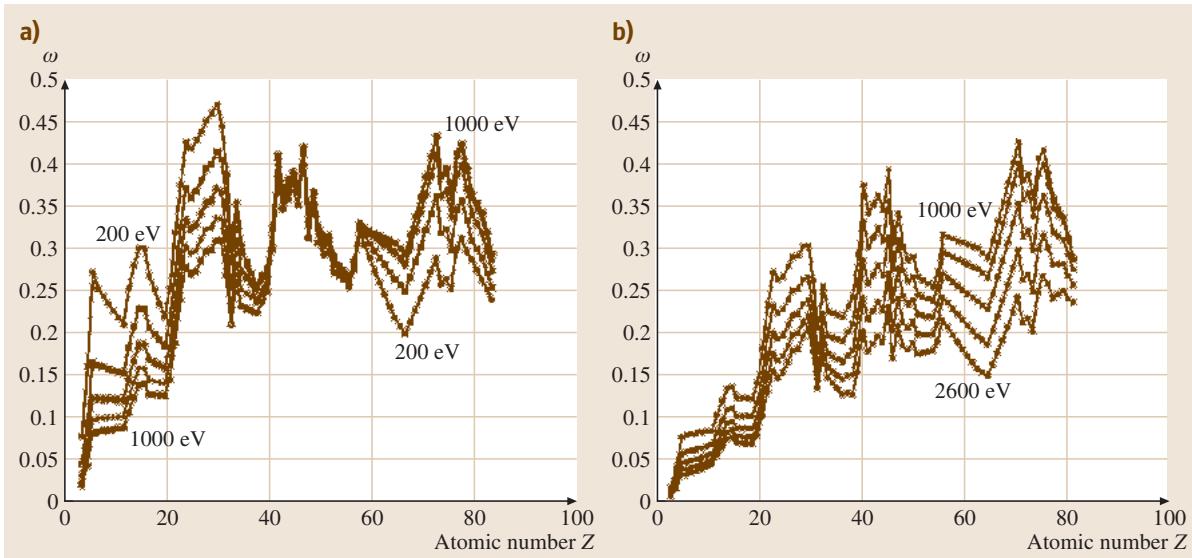 图1. 不同电子动能下,ω对原子序数Z的依赖关系:(a) 200至1000 eV区间,每隔200 eV;(b) 1000至2600 eV区间,每隔400 eV
图1. 不同电子动能下,ω对原子序数Z的依赖关系:(a) 200至1000 eV区间,每隔200 eV;(b) 1000至2600 eV区间,每隔400 eV
上述计算给出的是纯元素相对灵敏度因子(PERSFs)。然而,对于实际的定量分析,我们真正需要的是考虑了基体效应的平均基体相对灵敏度因子(AMRSFs)。如果直接使用PERSFs进行计算,忽略了相关的基体因子,所引入的误差范围可能在0.3到3倍之间。
过去,PERSFs和AMRSFs之间的差异并未得到充分认识。而且,实验数据大多来自化合物而非纯元素,因此本质上更接近AMRSFs。但这些数据缺乏能谱仪校准,且常常与PERSF的计算结果混合使用,导致参数定义不清。设备制造商不得不通过调整这些参数,以确保其设备在测试某些参考化合物时能给出“正确”的结果。
获取准确的定量结果,不仅需要正确的理论模型,更依赖于严格的实验操作和校准流程。这正是专业检测实验室的核心价值所在。 精工博研测试技术(河南)有限公司(原郑州三磨所国家磨料磨具质量检验检测中心),央企,国字头检测机构,专业的权威第三方检测机构,专业检测XPS定量分析,可靠准确。欢迎沟通交流,电话19939716636
测量厚度达8 nm的覆盖层是XPS的一项重要应用。对于厚度为d的纯A层覆盖在基底B上,其信号强度由以下公式描述:
I_A = I_A^∞ (1 - exp{-d / [L_A(E_A)cosθ]})
I_B = I_B^∞ exp{-d / [L_A(E_B)cosθ]}
在不考虑弹性散射的近似下,L_A值即为非弹性平均自由程(IMFP)λ_A。然而,Cumpson和Seah的研究表明,在存在弹性散射时,应使用衰减长度L_A替代λ_A,并且上述公式在θ ≤ 58°时有效。Seah和Gilmore的分析进一步指出,弹性散射导致:
L/λ = 0.979 [1 - ω (0.955 - 0.0777 lnZ)]
Jablonski和Powell的更精细计算也给出了相似的结果。
对于一般的薄膜,由于E_A和E_B通常不相等,直接从I_A和I_B求解厚度d并不容易,需要迭代计算。为此,Cumpson设计了“Thickogram”图来简化这个问题。
一个更巧妙的方法是利用XPS的化学态分辨能力。例如,在分析金属氧化物覆盖层时,与其使用氧峰和基底金属峰(存在表面吸附水汽的干扰),不如利用基底金属在氧化态(o)和元素态(e)的两个信号。这样做的好处是,两个信号的能量E_A和E_B非常接近,其差异可以忽略。此时,厚度d的计算公式简化为:
d = L_o cosθ ln(1 + R_expt / R_o)
其中,R_expt = I_o / I_e是实验测得的氧化态与元素态信号强度比,R_o = I_o^∞ / I_e^∞是无限厚氧化物与无限厚纯金属的信号强度比。R_o值可以通过计算得到,但为了精确测量d,强烈建议通过实验测量得到,并采用与后续分析相同的峰拟合方法。
利用该方法对硅片上的热生长SiO₂薄层进行厚度定量,已经在一项大型国际比对研究中得到了验证。该研究涉及与中能离子散射(MEIS)、卢瑟福背散射(RBS)、弹性背散射(EBS)、核反应分析(NRA)、二次离子质谱(SIMS)、椭圆偏振光谱、掠入射X射线反射(GIXRR)、中子反射(NR)和透射电子显微镜(TEM)等多种技术的比较。样品厚度范围为1.5 nm至8 nm,覆盖了超薄栅氧的应用范围。
为了在XPS中可靠地使用该公式,必须平均或避免由晶体衬底引起的衍射或前向聚焦效应。为此,研究人员定义了一种参考几何(RG):对于(100)表面,发射方向为距表面法线34°,方位角距[011]方向22.5°;对于(111)表面,发射方向为距表面法线25.5°,方位角在[101]方向。如果不采用这些特定取向,R_o值会偏小,导致计算出的厚度在薄膜区域出现系统性偏差。采用上述方法,可以获得极好的线性关系和与其他方法的高度相关性,使得L_o的校准精度可达1%以内,从而让XPS实现极高精度的厚度测量。
在该研究中,当其他技术的测量厚度d与XPS测得的厚度d_XPS进行关联时,多数方法呈现出d = m d_XPS + c的关系,其中存在一个偏移量c(见图2)。
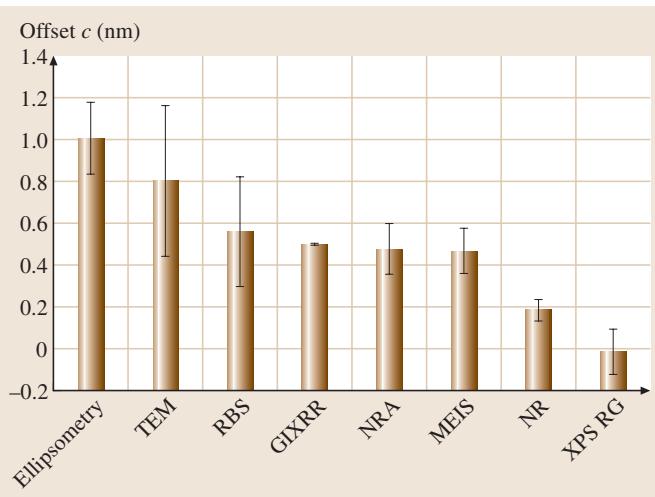 图2. 各技术与XPS测厚结果相比的偏移量c的平均值与标准差。注意一个原子层厚度约0.25 nm。
图2. 各技术与XPS测厚结果相比的偏移量c的平均值与标准差。注意一个原子层厚度约0.25 nm。
对于MEIS、NRA和RBS,这个偏移量代表了吸附的含氧物质(如水)的厚度,因为这些技术测量的是总氧量,而非仅限于SiO₂中的氧。0.5 nm的偏移量相当于一到两个单分子层的水。对于在空气中测量的椭偏仪,表面还有一层碳氢化合物和物理吸附水,使偏移量增加到约1 nm。TEM的偏移量原因尚不明确,可能源于对薄膜厚度定义的累积误差。GIXRR和NR的偏移量也源于表面污染,但在NR中影响较弱,未来或许能通过建模完全去除。
XPS结合离子溅射进行深度剖析,在原理上与AES深度剖析没有本质区别,但在实际操作中存在一些问题。
如果使用非单色化X射线源,其分析面积较大,要求溅射坑的面积也相应增大,这往往导致深度分辨率变差,因为要在大面积上维持平坦均匀的溅射深度非常困难。对于聚焦单色化X射线源,这个问题会得到缓解,但深度分辨率通常仍不及AES。
维持X射线源周围的良好真空环境,也使得人们不太倾向于使用原位的大面积离子枪,因为后者会带来较高的气体负载。
更根本的问题是,离子束的择优溅射会改变样品组分,这使得XPS测量化学态的核心优势大打折扣。因此,XPS深度剖析的应用不如AES广泛,但从原理上讲,它依然是一种可行的分析手段。如果您在实际工作中也面临类似的薄膜厚度检测或失效分析挑战,我们非常乐意与您一同探讨解决方案。












 首页
首页
 检测领域
检测领域
 服务项目
服务项目
 咨询报价
咨询报价
