
俄歇电子能谱(Auger Electron Spectroscopy, AES)技术,其核心在于利用一束高能电子束与样品表层原子的精妙互动。通常,一束能量在 2 至 20 keV(典型值为 5 或 10 keV)的电子束轰击样品表面,将表层原子的内层电子激发并打出,形成一个空穴。这个处于激发态的原子极不稳定,会迅速通过两种主要途径退激,回归稳态:
俄歇电子的动能 EA 携带了这三个关键能级的特征信息,其大小可近似由以下公式定义:
EA = E2 + E3 - E1
在这里,E1、E2 和 E3 这三个束缚能级的能量值通常取负值。弛豫效应会带来约 5 到 10 eV 的微小能量附加项,但上述公式足以让我们识别出射电子能量谱中的特征峰,进而鉴定除 H 和 He 之外的所有元素。
 图1:能级图示意发射过程:(a) 俄歇电子,(b) 光电子。阴影区域表示能级所参考的导带或价带。
图1:能级图示意发射过程:(a) 俄歇电子,(b) 光电子。阴影区域表示能级所参考的导带或价带。
现代AES仪器所用的电子束流通常在 5 到 50 nA 之间,可实现 20 nm 到 2 μm 的空间分辨率;在精密设计的仪器中,分辨率甚至可以优于 5 nm。
AES的分析范围也很有讲究。深度超过约 2.5 keV 的芯层,其电离截面很弱,导致产生的高能俄歇电子信号微弱,因此很少被用于分析。而在另一端,能量低于 50 eV 的俄歇电子又会被淹没在强烈的二次电子发射背景中。因此,用于分析的特征峰通常落在 50 eV 到 2500 eV 的动能区间内,这个窗口恰好覆盖了除 H 和 He 之外的所有元素。
AES之所以具备出色的表面特异性和灵敏度,其根本原因在于俄歇电子的“逃逸深度”。一旦俄歇电子在逸出过程中发生非弹性散射,就会损失能量,从特征峰中消失并融入背景信号。这个逃逸深度,即电子衰减长度 L,早期研究给出的经验公式为:
L = 538/EA2 + 0.41(aEA)0.5 (单位:单原子层)
其中,a3 是原子体积(单位:nm3)。以典型的 a 值 0.25 nm 计算,L 的范围从未满一个单原子层(低能端)到十个单原子层(高能端)不等。这决定了AES是一种极表面的分析技术。
下面这张表(表1)列出了一些关于表面化学分析(SCA)的ISO标准草案,涵盖了AES、SIMS和XPS等技术。
表1:ISO TC 201关于AES, SIMS和XPS的标准草案
| 编号 | ISO标准 | 标题(为节省空间已缩写) |
|---|---|---|
| 1 | TR 16268 | SCA – 离子注入表面分析标准物质 – 工作标准物质中保留面剂量的标准化程序 |
| 2 | 18115 Amd2 | SCA – 词汇 – 第2次修订 |
| 3 | 18117 | SCA – 分析前样品的处理 |
| 4 | TR 18392 | SCA – XPS – 确定背景的程序 |
| 5 | TR 18394 | SCA – AES – 化学信息的推导 |
| 6 | 18516 | SCA – AES和XPS – 横向分辨率的测定 |
| 7 | 20903 | SCA – AES和XPS – 峰强度的测定 |
| 8 | 22335 | SCA – 深度剖析 – 溅射速率的测量:使用机械式探针轮廓仪的网格复制法 |
| 9 | 22474 | SCA – AES和XPS中检测峰的方法指南 |
注:SCA=表面化学分析, SIMS=二次离子质谱, TR=技术报告, XPS=X射线光电子能谱。
图2展示了一张典型的俄歇电子能谱图,该研究旨在表征用于定量分析钢中磷元素晶界偏析的材料。为了获得这样的谱图,样品需要在谱仪的原位(in situ)断裂,以暴露新鲜的晶界表面进行分析。
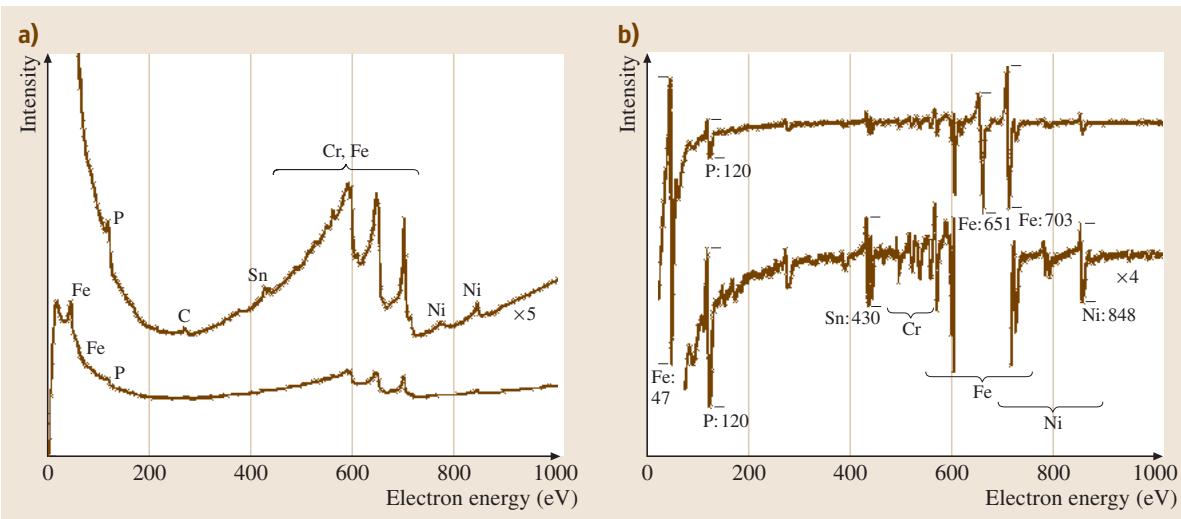 图2:(a) 直接模式和 (b) 微分模式下的低合金钢晶界断口俄歇电子能谱。该钢添加了0.056 wt.%的磷,并经过回火脆化热处理。样品在超高真空下冲击断裂,以防止暴露于空气中发生氧化。
图2:(a) 直接模式和 (b) 微分模式下的低合金钢晶界断口俄歇电子能谱。该钢添加了0.056 wt.%的磷,并经过回火脆化热处理。样品在超高真空下冲击断裂,以防止暴露于空气中发生氧化。
谱图中的峰同时以直接模式和微分模式进行了标注。从历史上看,能谱主要以微分模式呈现,因为这样可以可靠地测量像P这样的低能峰以及Cr等微量组分,这些峰往往出现在陡峭的背景斜坡上。由于缺乏普适有效的背景扣除方法,即使今天我们能以直接模式记录谱图,也常常需要通过数学程序(使用2至5 eV宽度的函数)对其进行微分处理。
谱图的形状和峰位可以查阅相关手册。AES中的跃迁通常采用X射线命名法。例如,如果公式(6.1)中的能级1、2、3分别是2p3/2、3p3/2和3d5/2亚层,则该跃迁记为 L3M3M5。对于从Ca到Cu的金属,由于M5处于价带中,常写作L3M3V。涉及价带的俄歇峰通常比仅涉及芯层的峰更宽,而涉及两个价带电子的峰则可能更宽。
峰的精确能量和形状会随化学状态而变化。涉及价带的跃迁受化学状态影响最大,例如,在氧化后可观察到高达6 eV的峰位移动和峰形分裂。图2中铁的三个主峰分别是位于600、651和700 eV的LCC、LCV和LVV峰,其中C代表芯层。在从Ti到Cu的元素序列中,LVV跃迁受氧化的影响最为显著,尤以Cr为最。
AES的两大早期应用是成像和结合惰性气体离子溅射的成分深度剖析。在成像应用中,分析人员通常满足于使用直接谱的峰强度,扣除一个外推的背景,使图像中每个点的亮度与峰高成正比。这在现代多检测器系统中既快速又简便。早期的系统则使用模拟微分谱的负向峰信号。这些用法简单实用,能快速确定待分析点,但对于表面形貌复杂的样品,或下方存在不同原子序数材料的薄层,图像衬度的解读会变得异常复杂。
在成分深度剖析中,通常使用能量为1至5 keV、电流密度为1至30 μA/cm²的氩离子进行溅射。剖析的对象可以是单原子层级别的,如图3(a)所示的钢中偏析的磷;也可以是较厚的膜层,如图3(b)所示的五氧化二钽(Ta2O5)认证标准物质(CRM)。深度和深度分辨率的量化是这里的关键因素,我们将在后面深入探讨。
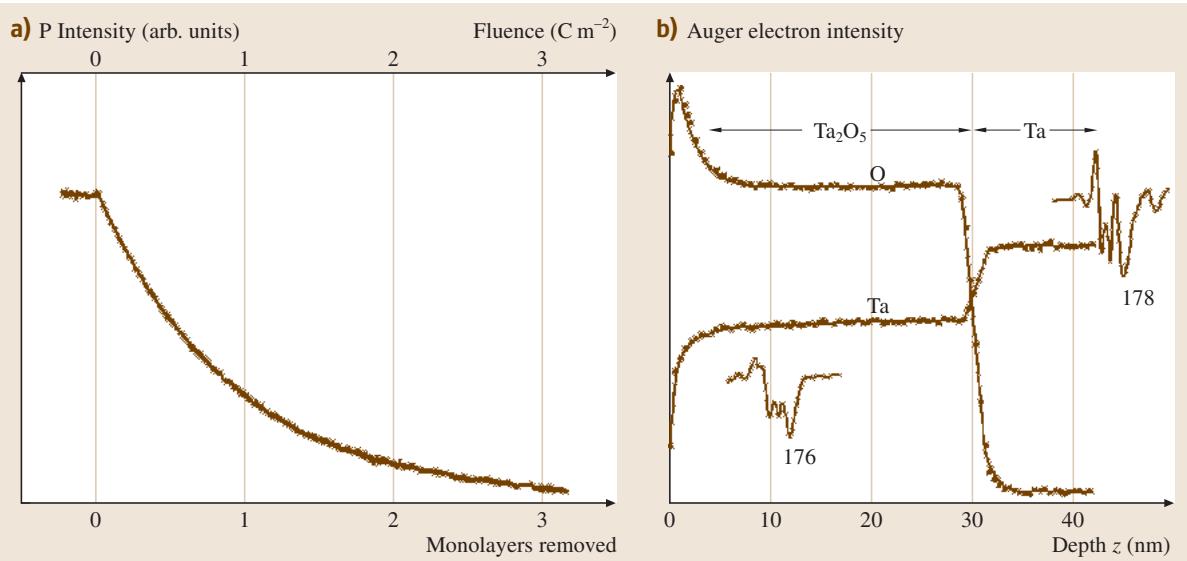 图3:使用氩离子进行AES深度剖析的微分信号图。(a) 来自图2(b)中低合金钢的120 eV处的P信号(3 keV离子溅射);(b) 来自28.4 nm厚的Ta2O5层的O和Ta信号(2 keV离子溅射)。钢样品在原位断裂后进行剖析,未接触空气;而Ta2O5层则在制备六个月后,一直保存在实验室空气中。
图3:使用氩离子进行AES深度剖析的微分信号图。(a) 来自图2(b)中低合金钢的120 eV处的P信号(3 keV离子溅射);(b) 来自28.4 nm厚的Ta2O5层的O和Ta信号(2 keV离子溅射)。钢样品在原位断裂后进行剖析,未接触空气;而Ta2O5层则在制备六个月后,一直保存在实验室空气中。
在许多AES应用中,样品是在仪器的超高真空环境中原位制备的,用户会遵循实验室或已发表文献中验证过的详细规程。然而,当样品来自外部,或者分析目标本身就是待深度剖析的覆盖层时,样品表面通常会带有污染物。这些污染物主要是碳氢化合物,会衰减所有待测信号。除非这些碳氢化合物是分析对象,否则最好将其去除。对于大多数材料,HPLC级别的异丙醇(IPA)是一种非常有效的溶剂,但需注意聚合物或其他可溶于IPA的材料。清洁表面不仅使分析更清晰、更准确,而且在深度剖析中,能去除许多低溅射产额的物质,避免后续剖析中深度分辨率的损失。样品随后可以存放在洁净的玻璃容器中,或直接进行分析。
关于如何收集和提供样品给分析人员的细节,可参考ISO 18117草案,而样品安装则可参考ISO 18116草案。样品安装的指导原则是,减少任何可能在真空系统中产生气体、污染待分析表面或导致绝缘材料局部充电的物质。
根据能谱仪的类型和用途,有两个ISO标准为校准能量尺度提供了程序。ISO 17973适用于为元素分析设计的中等分辨率系统(相对分辨率R ≤ 0.5%),无论是在直接模式还是微分模式(峰-峰微分宽度2 eV)下使用。ISO 17974则针对旨在进行元素和化学态分析的高分辨率能谱仪。两种标准都使用高纯度金属箔(如Cu、Au、Al),其表面通过轻微的离子溅射进行清洁。精确的峰能量由一个简单而准确的协议定义,然后与可溯源测量得到的表格值进行比较。
对于中等分辨率能谱仪,参考峰能量如表2所示,大多数仪器仅需Cu和Au箔。注意,能量值同时参考了真空能级和费米能级(括号内)。原则上,能量只能精确地参考费米能级,因为真空能级(谱仪真空中静止电子所处的能级)在谱仪内各点不同,会随烘烤和局部表面功函数而变。为方便起见,这些ISO标准和其他文献中采用了一个4.5 eV的约定值来表示费米能级与真空能级的差值。这里使用真空能级参考,是因为所有早期工作和手册都沿用了这一传统。
表2:参考动能Erefn (eV),参考真空能级(括号内为参考费米能级)
| 峰编号 n | 归属 | 直接谱动能 (eV) | 微分谱动能 (eV) |
|---|---|---|---|
| 1 | Cu M2,3VV | 58 (62) | 60 (64) |
| 2 | Cu L3VV | 914 (919) | 915** (920**) |
| 3 | Al KL2,3L2,3 | 1388 (1393) | 1390** (1395**) |
| 4 | Au M5N6,7N6,7 | 2011* (2016*) | 2021 (2026) |
| *对于低于6keV的束能且0.25% < R ≤ 0.5%,加1eV。 | |||
| **对于0.27% < R ≤ 0.5%,加1eV。 |
对于高分辨率能谱仪,真空能级的定义过于模糊,数据必须参考费米能级。这类仪器也常用于XPS分析,而XPS只使用费米能级参考,这增强了数据的一致性。高分辨率仪器同样使用Cu和Au,除非在谱仪量程限制在2 keV以下的特殊情况下,必须用Al替代Au,如表3所示。
表3:高分辨率谱仪动能标尺参考值 Erefno (eV) (参考费米能级)
| 峰编号 n | 归属 | Erefno (eV) |
|---|---|---|
| 1 | Cu M2,3VV | 62.37 |
| 2 | Cu L3VV | 918.69 |
| 3 | Al KL2,3L2,3 | 1393.09 |
| 4 | Au M5N6,7N6,7 | 2015.80 |
为达到必要的准确度,当分辨率R较差时(使用Au时R > 0.07%,或使用Al时R > 0.04%),需要对表格值进行校正,校正后的峰位 Erefn 为:
Erefn = Erefno + cR + dR2
系数c和d的值见表4,其中分辨率R(由ΔE/E给出)以百分比表示。
表4:分辨率较差时参考动能的校正系数
| 峰编号 n | 归属 | c (eV) | d (eV) |
|---|---|---|---|
| 1 | Cu M2,3VV | 0.0 | 0.0 |
| 2 | Cu L3VV | 0.2 | -2.0 |
| 3 | Al KL2,3L2,3 | -0.3 | -1.8 |
| 4 | Au M5N6,7N6,7: 5 keV n(E) | 0.0 | 0.0 |
| 5 keV En(E) | -0.3 | 4.4 | |
| 10 keV n(E) | -0.2 | 0.0 | |
| 10 keV En(E) | -0.1 | 0.0 |
这些ISO标准详细规定了信号水平、不确定度来源以及确保仪器在规定公差范围内保持校准的方法,以保证其适用性。
所有电子能谱仪都使用电子倍增器作为探测器,不幸的是,它们会随着使用而老化。这意味着,即使分析人员每次都使用一致的谱仪设置,所测谱图的绝对强度也会缓慢下降。虽然可以通过提高倍增器电压来弥补,但这又可能导致谱图中各峰的相对强度发生变化,从而影响定量分析结果。因此,分析人员必须理解探测器的行为,以维持测量的长期重复性。
对于脉冲计数系统,必须将倍增器增益(由电压控制)设置在一个合适的“坪区”,确保所有信号脉冲都被清晰地从背景噪声中分辨出来。NPL(英国国家物理实验室)推荐的程序是,将倍增器电压设置在比观察到50%饱和计数率(在约100 kc/s计数率下)所需电压高500 V的位置。这个设置能提供可靠的结果,并允许用户跟踪倍增器老化情况,以便在合适的时间更换。
正确设置探测器后,建立仪器强度响应的稳定性和重复性至关重要。对于AES,Cu L3VV峰与M2,3VV峰的强度比是一个有用的衡量标准,特别是使用峰-峰微分高度时。根据ISO 24236,通过足够高的计数(如每通道约2M计数),并采用特定的平滑方法,可以实现优于0.5%的重复性标准偏差。任何绝对强度或强度比的漂移都可能预示着源、分析器或探测器的不稳定性,需要进一步排查。
实验室间的比对研究表明,不同实验室获得的谱图形状存在显著差异,如果使用相同的相对灵敏度因子,定量结果可能相差两倍之多。这种差异主要源于探测器效率D(E)随时间的老化。D(E)随探测电子动能E变化,从零开始上升,在200-600 eV达到最大值,然后缓慢下降。
仪器的总响应函数(IERF)是探测器效率D(E)和谱仪传输函数T(E)的乘积:
IERF = T(E)D(E)
知道了IERF,我们就可以通过下式得到真实的能谱n(E):
n(E) = I(E) / IERF
其中I(E)是测得的谱图。为了校准IERF,研究人员使用带法拉第杯探测器的仪器化能谱仪,测量了Cu、Ag和Au多晶箔的绝对参考谱。基于这些参考谱,任何谱仪的IERF都可以被确定。已有软件系统被设计出来,方便用户根据自己测量的箔片数据进行自校准。使用三种箔片是为了评估三次独立IERF推导的分散性,从而计算平均IERF推导的重复性,其一致性可以优于1%。
若不进行严格的IERF校准,而仅仅依赖手册中的相对灵敏度因子和峰-峰微分强度进行定量,会引入巨大变数。研究表明,由于缺乏对IERF的控制以及微分调制能量选择的差异,已发表的灵敏度因子中,有一半的数值偏离平均值超过1.5倍。
理解灵敏度因子的基本推导,有助于我们欣赏某些操作方法的缘由。对于纯元素A的均质样品,涉及XYZ跃迁的俄歇电子强度IAXYZ∞ 可由下式计算:
IAXYZ∞ = γAXYZnAXσAX(E0)secα × [1 + rA(EAX,E0,α)]NAQA(EAXYZ) × λA(EAXYZ)cosθ (dΩ/4π)
这个复杂的公式包含了多个物理量:
这些参数的计算相当复杂,例如IMFP的计算就需要用到密度、价电子数、原子量等多个参数,不过已有免费软件可以辅助这个过程。
许多分析人员为了简化,使用一个非常基础的公式进行定量:
XA = (IA / IA∞) / Σi(Ii / Ii∞)
这里的 Ii∞ 是从手册中查到的纯元素相对灵敏度因子(PERSF)。这个方法虽然简单,但忽略了一个至关重要的因素:基体效应。
更精确的定量需要引入基体修正因子 FiM:
XA = [FAM(IAM / IA∞)] / Σi[FiM(IiM / Ii∞)]
其中 FiM 包含了样品基体M相对于纯元素i在原子密度、背散射、IMFP等方面的差异。FiM 的值可以在0.1到7之间变化,忽略它会带来巨大的误差。
显然,要精确计算这些矩阵因子,需要对基体有预先了解,这在实际分析中构成了一个“先有鸡还是先有蛋”的难题。如何跨越这一障碍,获得真正可靠的定量结果,考验着分析者的经验和理论功底。这正是专业检测实验室的核心价值所在。
精工博研测试技术(河南)有限公司(原郑州三磨所国家磨料磨具质量检验检测中心),央企,国字头检测机构,专业的权威第三方检测机构,专业检测表面成分分析,可靠准确。欢迎沟通交流,电话19939716636
最近的一项研究提出了一种巧妙的解决方案:用“平均基体相对灵敏度因子”(AMRSF)IAAv 来替代纯元素相对灵敏度因子IA∞。在计算IAAv时,原子内部的参数(如产额、电离截面)保持元素A的特异性,而原子外部的参数(如原子密度、背散射因子、IMFP)则使用一个“平均基体”的值。相关的计算方法已在ISO 18118中给出。
对于表面原子层不均匀的样品,定量分析变得更为复杂。但在AES中,有一个对冶金学家和催化研究者特别重要的特例:单原子厚度的偏析层。其覆盖度θA(以衬底s的堆积密度表示的单层分数)由下式给出:
θA = XA * [Ls(EA) / as] * cosθ
其中,XA是原子分数,Ls(EA)是电子在覆盖层中的衰减长度,as3是衬底原子的体积。许多早期甚至近期的工作混淆了覆盖度θA和原子分数XA的概念,导致了1到10倍的误差。
溅射深度剖析的基本原理是,使用惰性气体离子束(通常是1-5 keV的氩离子)逐层剥离样品表面,同时用AES监测剩余表面的成分变化。
对于暴露于空气的样品,剖析开始时会先去除表面的碳氢污染层,此时下方材料的信号会迅速上升,如图3(b)所示。如果样品是原位制备的,则不会有这个过程(如图3(a))。
当溅射到化合物层时,可能会发生“择优溅射”,即其中一种元素被优先溅射掉,导致测得的成分不再反映溅射前的真实成分。例如,在图3(b)的Ta2O5层中,氧被优先剥离,导致表面成分从Ta2O5变为近似TaO。要准确定量这种情况,最好的方法是在相同条件下溅射一个已知的参比层。
深度剖析的关键是深度分辨率Δz。早期研究发现,对于金属层,Δz会随着深度z的增加而恶化,大致遵循 Δz = kz0.5 的关系,这主要是由于溅射过程中形成了表面形貌。Zalar的一项重大改进是建议在溅射时旋转样品,这能极大地改善深度分辨率,即使对于棘手的多晶金属层也能获得5 nm的优异结果。
另一个关键是绝对深度z的测量。通常使用探针轮廓仪、光学技术或原子力显微镜(AFM)来测量溅射坑的深度。在浅层剖析中,一些细微效应变得重要:离子注入引起的初始膨胀、溅射产额达到平衡前的变化(约1nm深度)、以及活性样品在暴露于空气后坑底氧化引起的膨胀。这些都可能导致深度-时间关系曲线不经过原点。
当样品包含不同材料的叠层时,溅射速率会逐层变化,不能用单一速率进行深度换算。图4显示了不同元素在氩离子轰击下的溅射产额差异巨大。
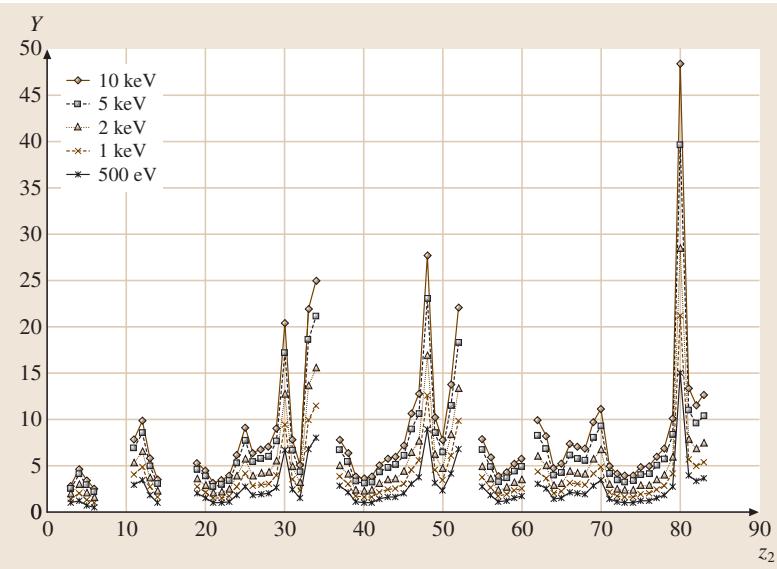 图4:在45°入射角下,使用不同能量的氩离子溅射各种元素时的计算溅射产额。
图4:在45°入射角下,使用不同能量的氩离子溅射各种元素时的计算溅射产额。
此时,需要通过计算或测量每层的溅射产额Y和离子束流密度J来确定深度d:
d = J * t * Y * a3 / e
其中t是溅射时间,e是电子电荷,a3是原子体积。尽管计算溅射产额的公式相当复杂,并且存在约10%的不确定性,但它为无法直接测量深度的情况提供了一种可行的替代方案。
在离子源的选择上,氩气(Ar)最为常用。如果Ar的俄歇峰与待测峰发生干扰,可选用氖(Ne)或氙(Xe)。一些分析人员偏爱Xe,认为它有时能带来更好的深度分辨率。
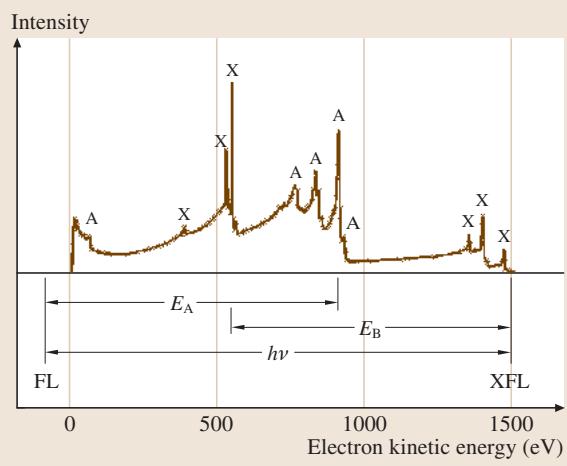 图5:使用非单色化Al X射线源获得的Cu的X射线光电子能谱。光电子峰标为“X”,俄歇电子峰标为“A”。图中也标示了Cu费米能级(FL)和光发射的费米能级电子(XFL)。
图5:使用非单色化Al X射线源获得的Cu的X射线光电子能谱。光电子峰标为“X”,俄歇电子峰标为“A”。图中也标示了Cu费米能级(FL)和光发射的费米能级电子(XFL)。
上一篇:超导材料:从物理原理到性能表征












 首页
首页
 检测领域
检测领域
 服务项目
服务项目
 咨询报价
咨询报价
