
现代核磁共振(NMR)技术早已超越了早期测量连续微波吸收的范畴。如今,我们通过探测自旋体系对一系列精心设计的微波脉冲序列的响应,并对采集到的振荡电流信号(即自由感应衰减,FID)进行傅里叶变换,从而获得高分辨率的核磁共振谱图。
为了精准操控和解读自旋行为,我们引入了旋转坐标系(Rotating Frame)这一强大的理论工具。在实验中,除了沿 ξ 轴施加一个强大的静态主磁场 B0 外,我们还会通过一个发射线圈,在垂直于 ξ 轴的 Χ 轴方向上,施加一个强度为 2B1、角频率为 ω 的交变磁场。这个交变场可以分解为两个强度为 B1、方向相反的圆周旋转磁场。
其中一个旋转磁场 B1 会对自旋产生两个关键效应:一是迫使原本各自进动的自旋实现同步;二是将它们的宏观磁化矢量从 ξ 轴(Z 轴)方向上“推”开。如果我们建立一个与该 B1 场以相同角频率 ω 围绕 ξ 轴旋转的坐标系(O-x’y’z’),那么在这个新坐标系中观察,自旋感受到的有效磁场将大大简化。它由两个恒定分量构成:一个沿 x’ 轴方向的静态场 B1,以及一个沿 ξ 轴方向的恒定场 B0 - ω/γn(其中 γn 是磁旋比)。
当施加的旋转场频率 ω 恰好满足共振条件,即 ω = ωL = γnB0 时,沿 ξ 轴的有效磁场分量 B0 - ω/γn 恰好为零。此时,在旋转坐标系中,自旋感受到的唯一磁场就是沿 x’ 轴的 B1 场。这个恒定的 B1 场会驱动宏观磁化矢量围绕 x’ 轴以一个较小的角频率 γnB1 进行旋转。这种将复杂的进动问题简化为简单旋转的矢量模型,为我们直观理解各种脉冲序列的效应提供了极大的便利。
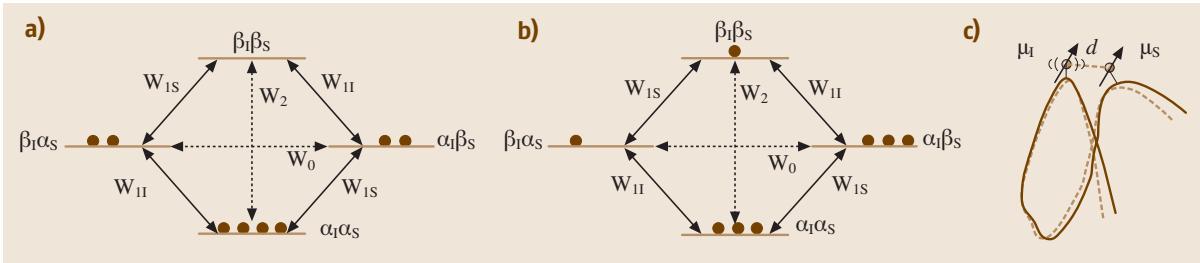 图1. 用于阐释核欧豪瑟效应(NOE)的能级图。NOE发生在空间上邻近的原子核之间。
图1. 用于阐释核欧豪瑟效应(NOE)的能级图。NOE发生在空间上邻近的原子核之间。
在NMR谱图中,化学位移的绝对大小与外磁场 B0 的强度成正比,而由 J-耦合产生的精细结构(裂分)大小则固定不变。这意味着,使用更高场强的磁体,可以有效“拉开”化学位移差异极小的重叠信号,从而显著提升谱图的分辨率(见图2)。对于蛋白质等生物大分子而言,其NMR谱通常表现出严重的信号重叠,因此提高分辨率是谱图解析的先决条件。目前,研究人员普遍采用超导磁体来获得极高的磁场强度,其场强通常以等效的 1H 共振频率来衡量,例如高达 800 MHz 甚至更高。
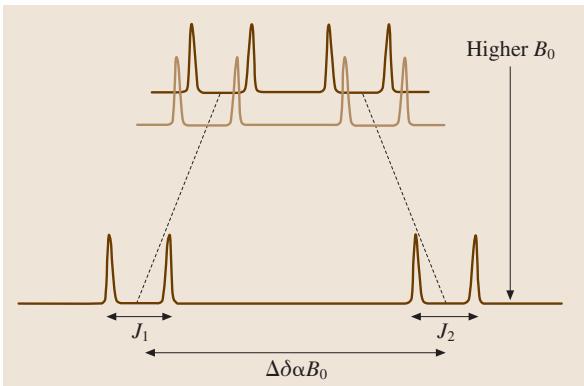 图2. 化学位移 Δδ 随磁场增强而增大,而自旋-自旋耦合常数 J1 和 J2 保持不变。使用高场强可有效提升NMR的分辨率。
图2. 化学位移 Δδ 随磁场增强而增大,而自旋-自旋耦合常数 J1 和 J2 保持不变。使用高场强可有效提升NMR的分辨率。
现代NMR技术的核心任务之一,就是无失真地分离如图3所示的复杂信号。除了采用高场磁体,设计精巧的一维脉冲序列也是解决问题的关键。
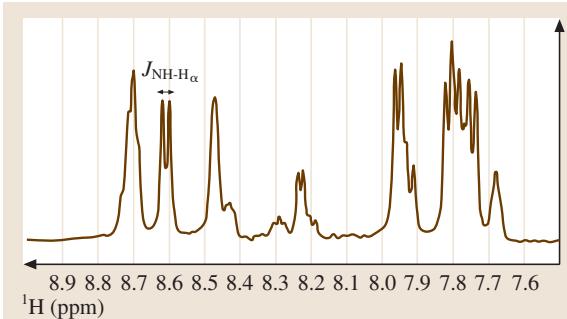 图3. 环状-GRGDSPA多肽的NMR谱图,展示了化学位移与J-耦合。
图3. 环状-GRGDSPA多肽的NMR谱图,展示了化学位移与J-耦合。
最基础的脉冲NMR实验可以用矢量模型清晰地展示(见图4)。
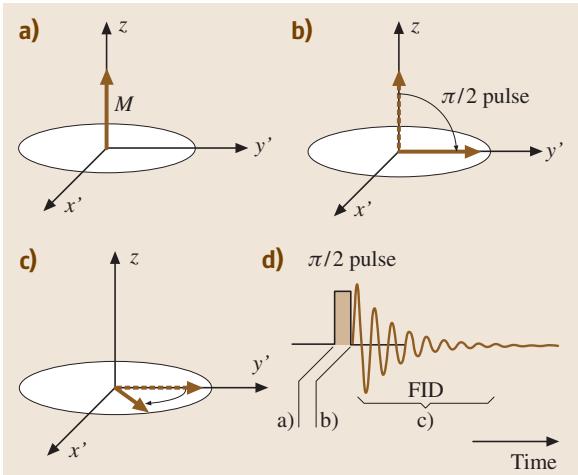 图4. (a)-© 旋转坐标系中矢量模型的磁化响应;(d) 自由感应衰减(FID)信号。
图4. (a)-© 旋转坐标系中矢量模型的磁化响应;(d) 自由感应衰减(FID)信号。
对这个FID信号进行傅里叶变换,即可得到频域的NMR谱图。如果样品中只有一种原子核,其谱图将是在其拉莫尔频率处的一个单峰(图5a)。
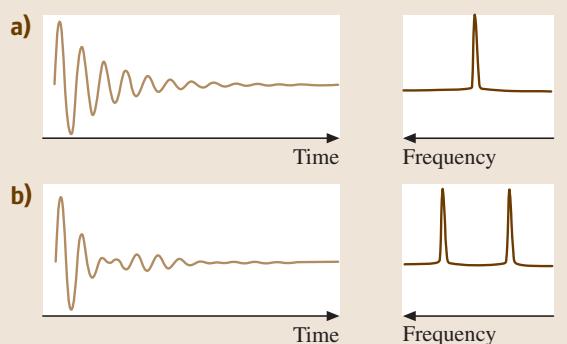 图5. (a, b) 从FID曲线(左)经傅里叶变换得到的NMR谱图(右)。
图5. (a, b) 从FID曲线(左)经傅里叶变换得到的NMR谱图(右)。
当体系中存在多种化学环境不同的原子核时,为了能用一个脉冲同时激发所有这些原子核,脉冲的持续时间 Δt 必须足够短(通常约 10μs),相应的 B1 强度则要足够大。这种脉冲被称为“硬脉冲”,其频率范围能覆盖所有目标核的化学位移。相反,持续时间长、强度弱的脉冲则称为“软脉冲”,它只能选择性地激发特定频率的原子核。
通用的一维NMR实验流程可概括为三个阶段:准备期(通过脉冲扰动自旋体系,使其脱离平衡态)、演化期(让体系在扰动后自由演化一段时间)和检测期(采集FID信号,探测演化期发生的变化)。
自旋去偶是一种强大的谱图简化与指认技术。假设原子核A和X通过J-耦合相互作用,导致彼此的信号都产生裂分。如果在检测A信号的同时,用一个强微波脉冲选择性地持续照射X核,X核的自旋态会发生极快地翻转。对于A核而言,它感受到的将是X核自旋态的平均效果,这使得J-耦合效应被消除,A的信号从裂分峰恢复为单峰。通过系统地对谱图中成对的裂分峰进行去偶实验,我们能够确认分子网络中相距3-4个化学键的J-耦合关系。
SPI是另一种揭示耦合关系的精妙方法,尤其适用于异核体系,例如 13C(A核)和 1H(X核)。在热平衡状态下,由于二者磁旋比 γ 的巨大差异,其能级布居数差别悬殊,表现为NMR信号强度的不同(图6a)。
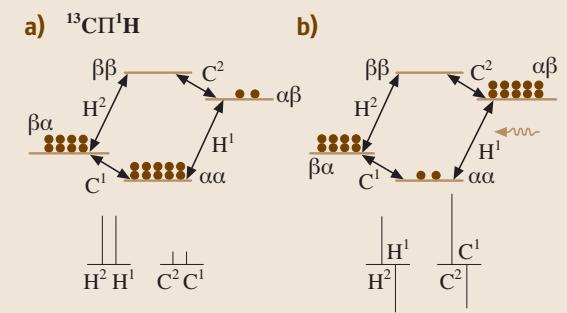 图6. (a, b) 异核选择性极化反转(SPI)。(a) 热平衡状态;(b) 当1H被选择性激发后。
图6. (a, b) 异核选择性极化反转(SPI)。(a) 热平衡状态;(b) 当1H被选择性激发后。
如果我们用一个软脉冲选择性地激发1H的某个跃迁,使其能级布居发生反转(图6b),13C的信号强度会发生剧烈变化,甚至出现相位相反的负信号。这种信号强度的变化直接证明了A核与X核之间存在耦合。与NOE效应不同,J-耦合导致的极化转移是瞬时发生的,这为我们区分J-耦合和偶极-偶极耦合提供了依据。此外,这类极化转移技术还能显著增强13C等低灵敏度原子核的信号。
对于小分子,一维NMR技术或许足以完成信号指认。但面对蛋白质这样谱线极度拥挤的大分子,必须借助更高级的手段。二维核磁共振(2D-NMR)应运而生,它将拥挤的信号在一个二维平面上展开,根据原子核之间的相互作用类型(如J-耦合或NOE)建立关联。
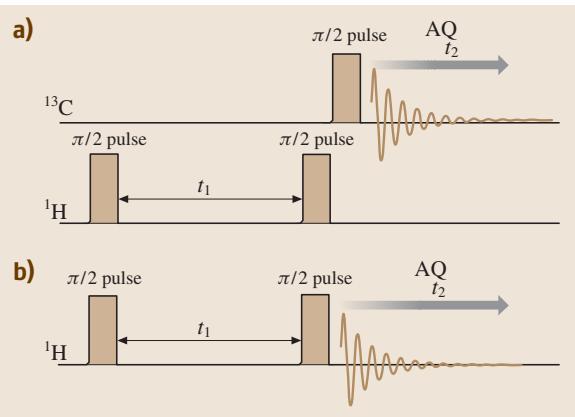 图7. 2D-NMR的脉冲序列示例。(a) 异核2D-NMR;(b) 同核2D-NMR (COSY)。
图7. 2D-NMR的脉冲序列示例。(a) 异核2D-NMR;(b) 同核2D-NMR (COSY)。
以一个异核(13C-1H)J-耦合体系为例(图7a),其脉冲序列与矢量模型演化如图8所示。在检测13C的FID信号之前,先对1H施加两个由时间 t1 隔开的90度脉冲。第一个脉冲将1H磁化矢量翻转至y’轴。在接下来的演化期 t1 内,该矢量以 ΩH = δH ± JCH/2 的频率在旋转坐标系中进动。第二个90度脉冲将这个演化后的矢量再次翻转。此时,1H磁化矢量在z轴上的投影分量大小为 Mcos(ΩHt1),这个分量的大小会通过J-耦合调制13C的能级布居,进而影响我们最终检测到的13C信号强度。
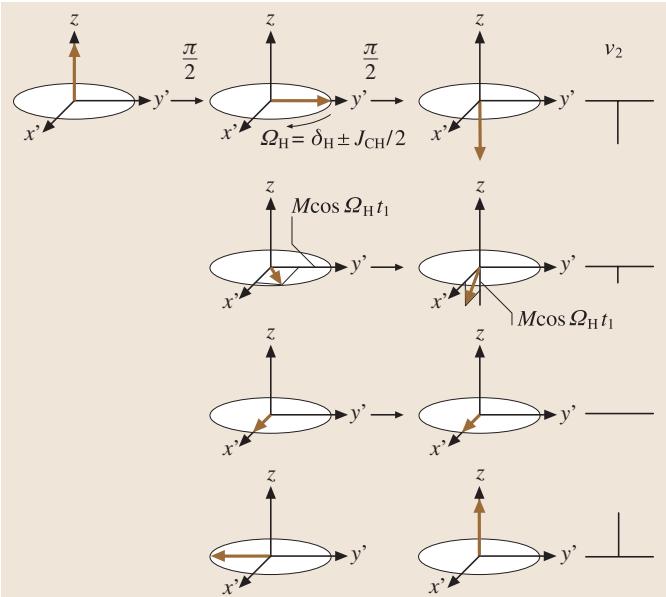 图8. 异核2D-NMR的矢量模型图解。
图8. 异核2D-NMR的矢量模型图解。
通过系统地改变演化时间 t1,并记录每次对应的13C信号强度,我们得到一个以 t1 为变量的信号振荡。对这个振荡信号进行第二次傅里叶变换(对 t1 维度),就能得到1H的频率信息。最终,我们获得一张二维谱图(图9),一个频率轴是13C的化学位移(ν2),另一个是1H的化学位移(ν1)。谱图上的“交叉峰”(off-diagonal peaks)清晰地表明了哪些13C和1H原子核之间存在J-耦合。这种方法极大地简化了复杂谱图的解析。
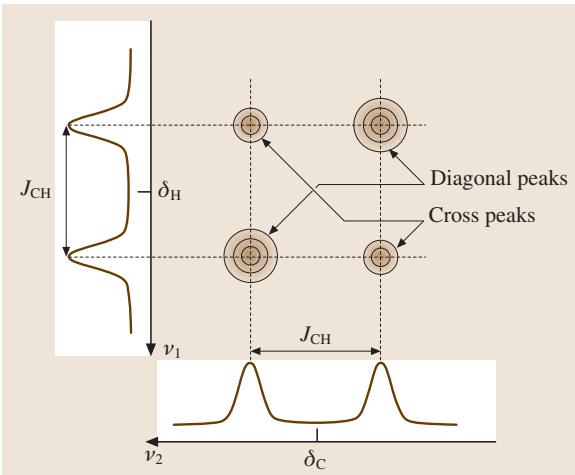 图9. 13C与1H J-耦合体系的示意性2D-NMR谱图。
图9. 13C与1H J-耦合体系的示意性2D-NMR谱图。
COSY(相关谱)是上述方法的同核版本,广泛用于寻找1H-1H之间的J-耦合关系(图7b)。其交叉峰表明两个质子通过化学键相连(通常在3-4个键之内)。图10展示了图3中多肽的COSY谱图,其交叉峰网络清晰地揭示了氨基酸残基内部的自旋系统。
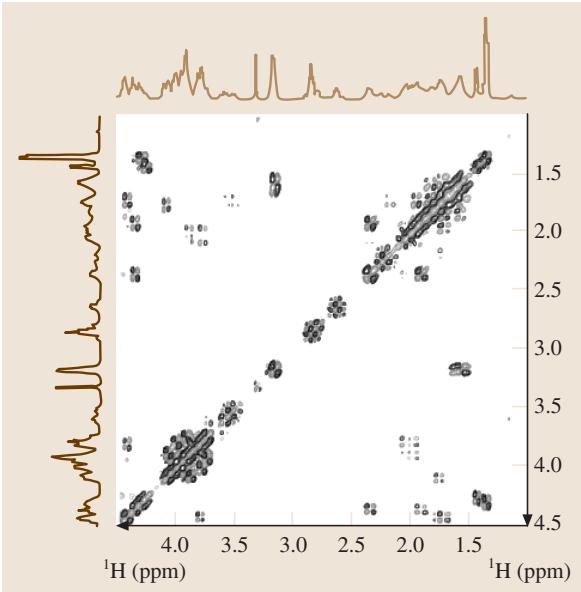 图10. 环状-GRGDSPA多肽的DQF-COSY谱图。
图10. 环状-GRGDSPA多肽的DQF-COSY谱图。
NOESY(核欧豪瑟效应谱)则是在演化期和检测期之间增加了一个“混合期”(mixing time, tm)(图11)。在混合期内,由于NOE效应(偶极-偶极相互作用),空间上邻近(通常< 5Å)的原子核之间会发生磁化交换。其二维谱图(见图12矢量模型)上的交叉峰直接反映了原子核在空间上的接近程度,这对于确定大分子的三维构象至关重要。
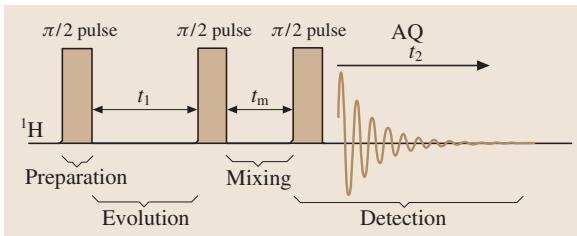 图11. NOESY实验的脉冲序列。
图11. NOESY实验的脉冲序列。
获取高质量的NMR数据对样品有一定要求。由于NMR信号强度与自旋数量成正比,通常需要毫克级别或浓度在1mM以上的样品才能获得足够的信噪比。
此外,谱图的质量也受多种因素影响。自旋量子数 I > 1/2 的原子核因强烈的四极矩相互作用,谱线通常很宽,不适合高分辨率研究。即便对于 I = 1/2 的核,磁场不均匀性也会导致FID信号快速衰减和谱线展宽。虽然自旋回波等技术可以部分克服磁场不均匀性,但由分子各向异性屏蔽导致的谱线展宽仍然是棘手问题。对于在溶液中快速运动的小分子,这种各向异性会被平均掉,得到尖锐的谱峰(运动窄化效应)。然而,对于运动缓慢的大分子,该效应不明显,导致谱图分辨率下降。
因此,对缺乏运动性的大分子进行NMR分析极具挑战性。尽管发展了多种技术来应对,但目前通过NMR进行结构解析的分子量上限大约在 3 × 104 Da 左右。这与X射线衍射技术形成了鲜明对比,后者原则上可以解析像核糖体(分子量 4 × 106 Da)这样巨大的分子复合物,前提是能获得高质量的单晶。要获得一张信噪比高、结果可靠的图谱,对样品制备、设备参数配置都有极高要求。这正是专业检测实验室的核心价值所在。
精工博研测试技术(河南)有限公司(原郑州三磨所国家磨料磨具质量检验检测中心),央企,国字头检测机构,专业的权威第三方检测机构,专业检测复杂体系NMR谱图分析,可靠准确。欢迎沟通交流,电话19939716636












 首页
首页
 检测领域
检测领域
 服务项目
服务项目
 咨询报价
咨询报价
