
在微观尺度上,绝大多数固体材料都展现出化学成分分异的微观结构,其特征尺寸覆盖了从微米到纳米的广阔范围。许多宏观尺度下的物理、生物乃至技术过程,其本质都受控于微观尺度上发生的化学变化。电子探针X射线显微分析仪(Electron Probe X-ray Microanalyzer, EPMA)正是一种为探究这一微观世界而生的强大分析工具。它基于扫描电子显微镜(SEM)的构架,利用一束精细聚焦的电子束激发样品,使其发射出具有元素特征的X射线,从而实现对微区成分的精准解读。
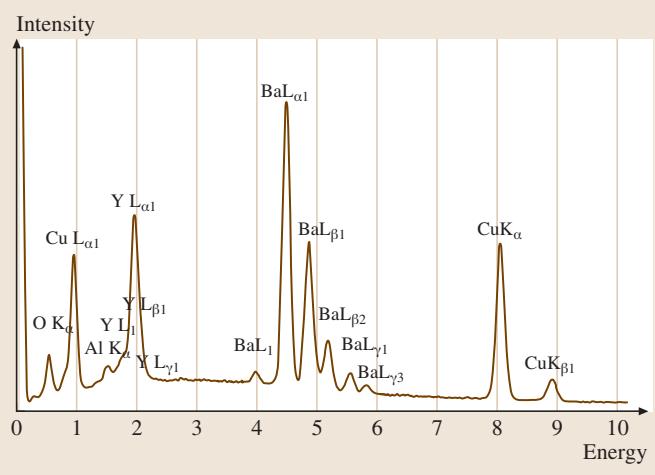
图1. 对含微量铝的 YBa2Cu3O7-x 单晶进行能量色散谱(EDS)分析。电子束能量为 20keV。
根据样品的具体成分、入射电子束的能量、特征X射线的能量以及分析条件的不同,EPMA的分析区域在横向和深度上的尺寸可在 50nm 到 5μm 之间变化。
EPMA/SEM 系统能够对主量、次量乃至痕量元素进行定量分析,其质量分数检出限可低至约 10-5,但对氢(H)、氦(He)和锂(Li)等超轻元素无能为力。该技术通常被视为一种无损检测方法,最适用于经过金相抛光的平整样品。得益于SEM的灵活性,EPMA的应用也拓展到了粗糙表面、颗粒、基底上的薄层乃至无支撑薄膜等特殊情形。此外,SEM本身集成的形貌成像和结构晶相学功能,使得在微米至纳米尺度上表征样品的地形、表层、横向成分变化、晶体取向以及磁场和电场分布成为可能。
在X射线探测方面,两种不同类型的谱仪被广泛使用:能量色散谱仪(Energy-Dispersive Spectrometer, EDS)和波长色散谱仪(Wavelength-Dispersive Spectrometer, WDS),后者亦称晶体衍射谱仪。这两种谱仪的特性形成了完美的互补关系:一方的弱点恰好是另一方的强项。因此,它们常常被集成在同一台电子束仪器上协同工作。近年来,硅漂移探测器(Silicon Drift Detector, SDD)的出现,更是将EDS的输出计数率提升至 100kHz 到 500kHz 的范围。图1展示了从一个多组分样品 YBa2Cu3O7 获得的典型EDS谱图,体现了其宽泛的能量覆盖范围。而图2则直观对比了EDS和WDS对镝(Dysprosium)元素L系X射线的分析结果,WDS在谱图分辨率上的显著优势一目了然。表1详细对比了这两种谱仪的关键性能参数。
定性分析的目标是识别谱图中的特征峰分别对应哪些元素。对于主量元素(质量分数 > 0.1%),这一过程通常直接明了。然而,当面对次量(0.01% - 0.1%)和痕量(< 0.01%)元素时,挑战便随之而来。特别是使用EDS分析时,如果次量或痕量元素的特征峰与主量元素的峰位非常接近(能量差 < 100eV),就会产生严重的谱峰重叠。这种干扰需要通过峰解谱技术来解决,对于原子序数较低的轻元素(Z < 18)尤其如此,因为它们可能只有一个峰能被EDS分辨。因此,任何由计算机软件自动完成的EDS定性分析结果,都必须经过经验丰富的人工审核以确保其准确性。
WDS凭借其卓越的谱图分辨率,通常能在这种情况下清晰地分离主量/次量或主量/痕量元素的谱峰。同时,WDS也不易受EDS中常见的谱图伪影(如堆积峰和逃逸峰)的影响。不过,使用WDS时也需格外小心,要避免将布拉格衍射方程中的高阶衍射(n = 2, 3, 4, …)误判为来自其他元素的特征峰。
表1. EDS与WDS X射线谱仪特性对比
| 特性 | EDS (半导体探测器) | WDS |
|---|---|---|
| 能量范围 | 0.1–25 keV (Si) 0.1–100 keV (Ge) |
0.1–12 keV (4 块晶体) |
| MnKα处的分辨率 | 130 eV (Si); 125 eV (Ge) | 2–20 eV (取决于能量和晶体) |
| 瞬时能量覆盖 | 全范围 | 分辨率宽度,2–20 eV |
| 死时间 | 50 μs | 1 μs |
| 接收立体角 (球面度) | 0.05 – 0.2 | 0.01 |
| 量子效率 | ≈ 100% (3 keV 至 15 keV, Si) | < 30%, 随能量变化 |
| 最大计数率 (EDS) | ≈ 3 kHz (最佳分辨率) ≈ 30 kHz (mapping) |
100 kHz (单一能量) |
| 最大计数率 (SDD) | ≈ 15 kHz (最佳分辨率) ≈ 400 kHz (mapping) |
|
| 全谱采集时间 | 10 s 至 200 s | 600 s 至 1800 s |
| 突出优势 | 可同时查看全谱,利于在所有位置进行定性分析 | 能解决谱峰干扰;脉冲响应快,适合成分面分布分析 |
实现精准的定量分析,其过程可分解为三个核心步骤:(1) 提取谱峰净强度;(2) 标准化处理;(3) 基体效应校正。
对于EDS分析,首先需利用背景模型或数学滤波器扣除谱图的背景。随后,采用多元线性最小二乘法(MLLSQ)进行峰解谱。MLLSQ方法要求为每一种待测元素建立一个不受其他元素干扰的、在该仪器上测得的标准峰形模型。对于未知样品中由多个元素贡献构成的重叠峰区,通过线性组合所有相关元素的参考峰形来构建一个合成谱,并将其与实测谱进行比较,直到基于卡方检验等统计学标准,逐通道地找到最佳匹配。
而对于WDS分析,其高分辨率通常足以分离开谱峰间的干扰,因此主要问题在于背景的扣除。由于在WDS单峰的窄能量窗口内,背景强度呈线性变化,因此可以通过在峰两侧测量两个背景点并进行线性内插,来实现精确的背景校正。
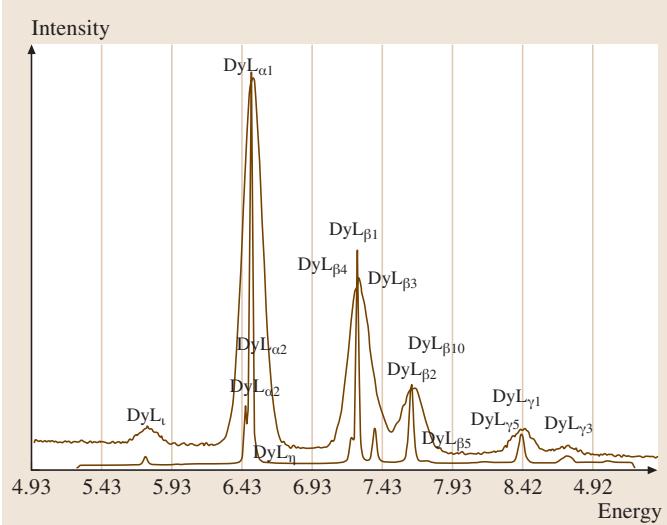
图2. 在20keV束流能量下,EDS与WDS对镝元素L系X射线的分析结果对比。
准确的电子探针定量分析,其基础是测量未知样品中某元素的X射线峰强度与标准样品中相同谱线强度的比值。所有测量都必须在相同的束流能量、已知的电子剂量(束流 × 时间)和谱仪效率下进行。这个比值被称为 k-值,它近似正比于该元素在样品和标样中的质量浓度比:
IA,样 / IA,标 = k ≈ CA,样 / CA,标
标准化步骤有效地消除了探测器效率的影响,并减弱了许多用于基体校正的物理参数所带来的不确定性。EPMA的一大优势在于其所需标样体系的简洁性。通常,纯元素以及那些在电子束轰击和真空环境下不稳定的元素的简单化学计量化合物(例如,用黄铁矿 FeS2 作为硫的标样)就足够了。这是一个巨大的便利,因为制备在微米尺度上均匀的多元素混合物,由于相分离等问题,通常是相当困难的。
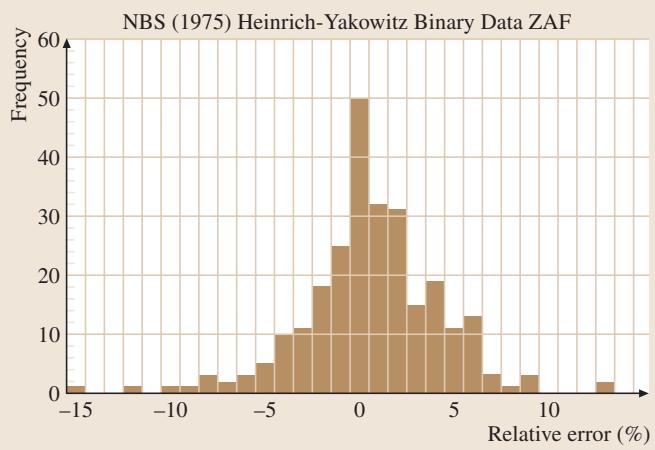
图3. 采用纯元素标样分析二元合金的相对误差分布。图中数据由美国国家标准局ZAF程序进行基体校正,采用WDS采集,测量精度通常为0.3%相对标准偏差。
k-值与浓度比(CA,样 / CA,标)之间的关系并非严格相等,这源于基体效应或称元素间效应的作用。也就是说,样品中B元素的存在会改变A元素X射线的产生、传播和被探测到的强度。幸运的是,这些基体效应的物理根源已被充分理解。通过结合基础物理原理和大量经验测量,科学家们发展出了一套乘法校正因子,分别对应原子序数效应(Z)、吸收效应(A)和荧光效应(F):
CA,样 / CA,标 = k · Z · A · F
显然,Z、A、F这三种基体效应都强烈依赖于待测样品的整体成分,而这恰恰是我们希望求解的未知量。因此,基体效应的计算必须通过迭代的方式进行。首先,使用测得的k-值作为样品组成的初始估计值,通常将其归一化:
Ci,1 = ki / Σki
其中 i 代表每个被测量的元素。然后,利用这组初始浓度值来计算第一轮的基体校正因子,进而计算出预测的k-值。将预测的k-值与实验测得的k-值进行比较,如果两者在设定的误差范围内一致,则计算终止;否则,重复此循环。通常,在三次迭代内即可收敛到最终结果。
在过去的几十年里,这种基体校正程序通过使用各种成分已知的微观均匀材料(如合金、矿物、化学计量二元化合物等)作为“未知”测试样,得到了反复的验证。图3展示了针对二元合金分析的一个典型相对误差分布情况,其结果的准确性可见一斑。要获得一张信噪比高、结果可靠的图谱,对样品制备、设备参数配置都有极高要求。这正是专业检测实验室的核心价值所在。 如果您在实际工作中也面临类似的材料微区成分分析挑战,我们非常乐意与您一同探讨解决方案。
精工博研测试技术(河南)有限公司(原郑州三磨所国家磨料磨具质量检验检测中心),央企,国字头检测机构,专业的权威第三方检测机构,专业检测合金元素定量分析,可靠准确。欢迎沟通交流,电话19939716636
将X射线微区分析信息以图像形式呈现,是一种极其强大的数据展示方法。这种被称为成分面分布图或元素分布图(Mapping)的技术,能够直观地描绘出各元素组分的空间分布。这些面分布图可以与提供形貌信息的SEM图像同步记录。
实现过程是,当电子束扫描样品表面的每一个像素点时,记录下WDS、EDS或SDD在对应于目标元素特征峰的特定X射线能量范围内的数字输出信号。最高级的成分面分布技术,甚至会在图像的每个像素点上采集一个完整的能谱,或至少是多个能量窗口的强度数据。这些海量的谱学数据随后经过背景扣除、谱峰解谱、标准化和基体效应校正等一系列处理,以实现真正的定量分析。最终生成的面分布图,实际上是样品局部浓度的记录。因此,当显示图像时,其灰度或色标是与元素浓度直接关联的。图4展示了对一种铝镍合金进行成分面分布分析的实例。

图4. 镍铝(Ni-Al)合金的成分面分布图(Ni、Al、Fe)和背散射电子(BSE)图像。图像揭示了复杂的微观结构,其中次量元素铁以非连续相的形式偏聚。












 首页
首页
 检测领域
检测领域
 服务项目
服务项目
 咨询报价
咨询报价
